烟草优质品种花全长cDNA文库的构建和质量鉴定 
2.福建省烟草专卖局烟草农业科学研究所, 福州, 350003
 作者
作者  通讯作者
通讯作者
《分子植物育种》网络版, 2011 年, 第 9 卷, 第 40 篇 doi: 10.5376/mpb.cn.2011.09.0040
收稿日期: 2010年11月18日 接受日期: 2011年02月28日 发表日期: 2011年04月08日
曾建斌等, 2011, 烟草优质品种花全长cDNA文库的构建和质量鉴定, 分子植物育种 Vol.9 No.40 (doi: 10.5376/mpb.cn.2011.09.0040)
以优质烤烟品种翠碧一号花为材料,CTAB法提取总RNA,按经修改的CreatorTM SMARTTM cDNA Library Construction kit的方法,成功构建了花全长cDNA文库。经鉴定,初级文库库容为4.05×106个克隆,重组率达到95%,插入片段集中在1 000bp~2 500 bp之间,平均长度约为1 400 bp,经扩增后的文库滴度为6.75×109 pfu/mL。随机挑取24个克隆进行测序,获得有效序列22条,利用Blast2GO软件进行初步分析,其中18条序列具有不同数量的功能注释。综合表明,所构建的文库质量高,可用于筛选花发育重要功能基因,这为揭示烟草花发育机理和烟草遗传改良提供了物质基础。
花器官是植物生殖过程中重要的功能器官,它的变异或败育对植物繁衍、种群扩散和农业生产将带来不利影响(华志明, 1998)。了解植物开花行为是农学和园艺生产上极具经济意义的课题。长期以来,对花的研究多限于形态描述以及开花生理方面,随着分子生物学技术突飞猛进的发展,从分子水平揭示花发育机理已经是植物分子生物学的活跃研究内容。
通过对双子叶模式植物拟南芥和金鱼草花器官突变体的研究,初步揭示了花器官发育的遗传控制机理,并建立了花器官发育ABC模型(崔荣峰和梦征, 2007)。该模型认为决定被子植物花各个器官发育的基因共分为A、B、C三类。随着研究的深入,克隆出的花同源异型基因数量不断增加,进而又发现了D、 E两类基因。这样经典的ABC模型就发展成为ABCDE模型(丛楠等, 2007)。这些花器官发育相关的同源异型基因,大都属于MADS-box基因家族,该家族成员作为一类转录调控因子在植物花发育调控中发挥着重要作用,到目前为止,已相继克隆出大量该基因家族成员(Kramer et al., 2004; Guo et al., 2007; 韩利涛等, 2010)。利用转录因子的调节功能,还能对开花期和花型进行调节,这在生产中具有重要意义。如拟南芥中的AP1基因转化番茄花期提前,出现自然整枝的现象,果实心室数目增多(王翊等, 2010)。通过转录因子调节花粉育性,可以产生不育的花粉,创造不育系,简化育种程序,对杂交育种等生产实践有重要意义。除此之外,现在已经证明miRNA在植物开花时间调控及花器官发育过程中也起着不可替代的作用(侍婷等, 2010)。
作为模式植物的拟南芥、矮牵牛、金鱼草、水稻和杨树花发育都有较多研究,而烟草(Nicotiana tabacum) 作为另一种重要的模式生物,但对其花发育的研究甚少。烟草是四倍体,基因组大而复杂,从功能基因组学出发发掘研究烟草花发育功能基因,有利于高效揭示花发育机理;另外,研究烟草花发育分子机理有助于补充对花发育的分子机理的认识,也必将对烟草遗传改良和杂种优势利用有意义。本文利用SMART (switching mechanism at 5'end of RNA transcript)技术(Lin et al., 2004; 王家保等, 2009 ; Li et al., 2010),成功构建了福建优质烤烟品种花不同发育时期的cDNA文库,为高效克隆和研究花发育功能基因奠定了基础。
1结果与分析
1.1总RNA的提取鉴定
高质量的RNA是构建高质量文库的前提。本研究提取的总RNA (见图1),用微量紫外分光光度计测A260/A280=1.82表明所提RNA较纯。从图中可以看出总RNA的28S rRNA和18S rRNA两条带清晰,且28S条带亮度约为18S的两倍,隐约可见5S rRNA,没有基因组DNA污染。以上结果表明,所提RNA没有降解,完整性较好,可以进行文库构建的后续工作。
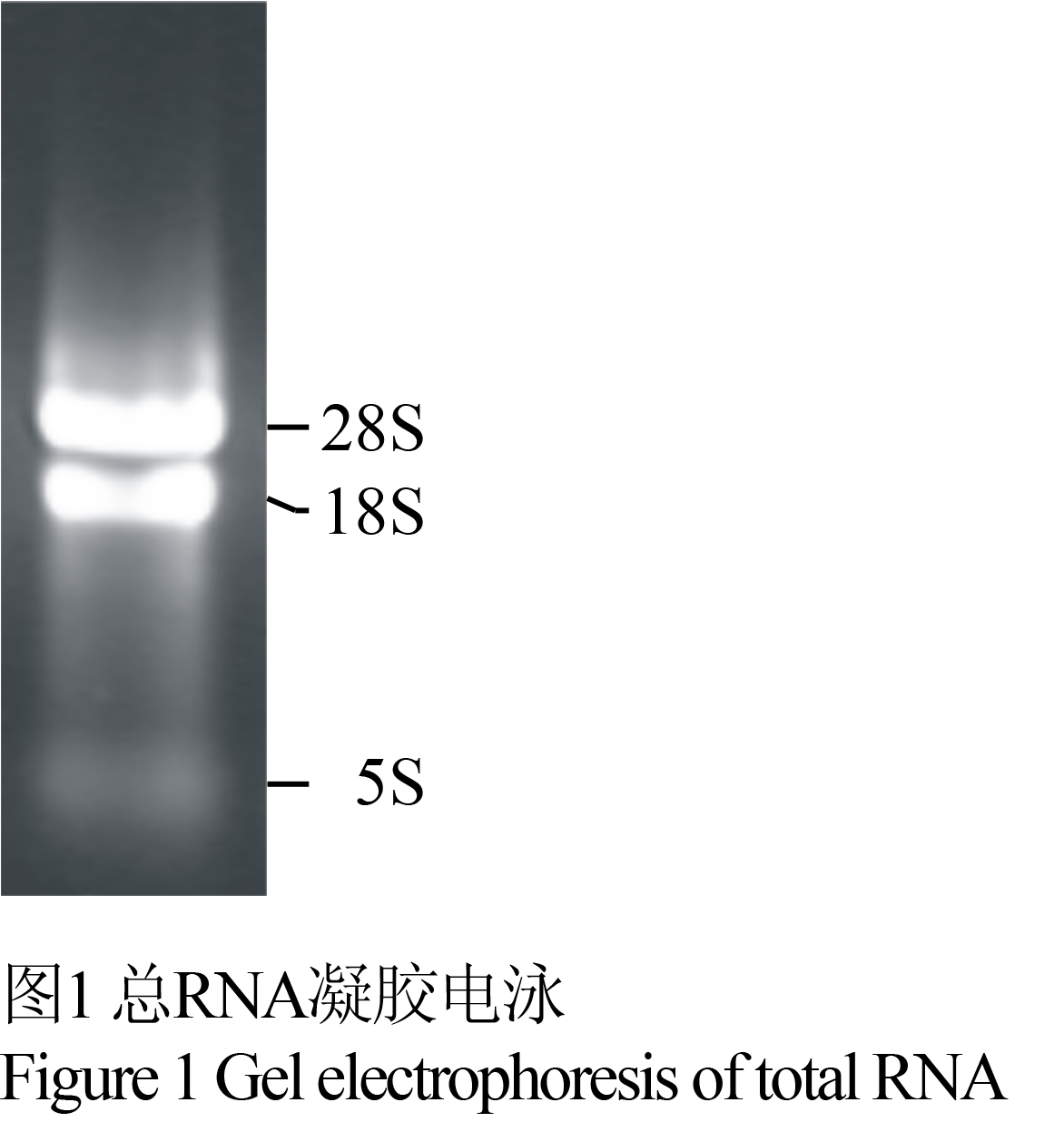 图1 总RNA凝胶电泳 Figure 1 Gel electrophoresis of total RNA |
1.2双链cDNA合成结果
总RNA逆转录得第一链cDNA,然后以抑制PCR方法合成双链cDNA (见图2),从图中可以看出双链呈弥散状分布,最高片段超过2 000 bp,信息较为完整,表明双链cDNA可用于连接和转化。
.png) 图2 双链cDNA凝胶电泳 Figure 2 Gel electrophoresis of double strand cDNA |
1.3文库质量鉴定
1.3.1文库库容量及滴度测定
原始文库菌液稀释涂板后,计算克隆数,按照cDNA文库容量计算公式:库容量=每μL文库菌液所含的菌落数×文库菌液总体积,得到初级文库库容为4.05×106;扩增文库滴度参照文库构建试剂盒CreatorTM SMARTTM cDNA Library Construction Kit说明书上的计算公式:文库滴度cfu/mL=菌落数×稀释倍数×103,得到扩增文库滴度为6.75×109 pfu/mL,可用于长期保存。
1.3.2文库重组率及插入片段鉴定
随机从板上挑取20个克隆,用载体通用引物M13F和M13R引物PCR扩增,1%的琼脂糖凝胶电泳检测(见图3)。如图3所示,在20个菌液PCR结果中,除泳道16外,剩余19个都有条带,重组率为95%。片段大小大都集中在1 000~2 500 bp之间,平均长度约为1 400 bp。
图3 随机挑取20个菌落的PCR检测 Figure 3 PCR detection of randomly selected 20 colonies |
再次从板上随机调取30个克隆,摇菌,提质粒后,用限制性内切酶SfiI酶切后,经1%的琼脂糖凝胶电泳检测(见图4)。如图4所示,插入片段大小与菌液PCR结果基本吻合,进一步验证了文库的质量。
.png) 图4 随机挑取30个菌落SfiI酶切 Figure 4 SfiI digestion of randomly selected 30 colonies |
1.4测序结果初步分析
测序获得有效序列为22条,按Blast2GO默认的参数运行完后,根据序列相似性分布自动输出物种相似性分布统计图如(图5),最高的为水稻和葡萄,紧接为拟南芥、毛果杨,烟草排在第五位。由于前四种植物基因组都相继破译,所拥有的基因资源丰富,而烟草基因组庞大而复杂,这就为其基因组的破译带来困难。因此,构建cDNA文库,从转录组研究烟草基因是十分必要的。
.png) 图5 物种相似性分布 Figure 5 Catelog of species distribution |
分别从分子功能、细胞组件、生物过程类别对序列进行GOs功能注释, 22条序列中具有注释的序列为18条(表1),剩余4条为未知功能或无功能的基因序列。在这些注释的功能中,参与了不同的细胞器组件,涉及到生物体内不同的代谢过程,如糖酵解、光合作用、脂肪酸代谢、核酸代谢等重要的生物过程,既有物质代谢过程,又伴随能量代谢过程。通过这些有限的基因功能注释结果可以预测,文库中信息量较为丰富,获得候选基因是可行的。
 表1 Blast2GO具有GOs功能注释的18条序列 Table 2 Eighteen sequences with the functional annotation by Blast2GO |
2讨论
SMART 法由Clontech公司创建,具有简单、快速等优点,目前被许多学者广泛运用于文库的构建。常规的SMART法步骤中,双链cDNA经过蛋白酶K消化和SfiI酶切后,较常用的是cDNA分级分离法收集片段,用于连接。往往构建的文库插入片段大都集中在1 000 bp左右,大片段占的比例很小。由于不同长度的cDNA片段在连接反应中的连接速度不同,短片段优先连接,长片段连接效率则低,导致了长片段cDNA克隆较少。本研究将琼脂糖凝胶电泳切胶回收的方法引入,取代cDNA分级分离法,构建的文库经鉴定大片段的比例明显提高。
cDNA文库的质量具体反映在两个方面(朱碧云, 2009):一是文库的代表性,可用一个量化的指标来衡量,即文库的库容量。本研究所构建的原始cDNA文库具有的库容为4.05×106个独立重组子,为满足筛到低丰度mRNA的要求,一般cDNA文库构建要求其库容不小于1×106 cfu/mL,本研究构建的cDNA文库完全达到了这个库容要求。二是重组cDNA片段的大小。本研究所构建的文库插入片段集中在1 000-2 500 bp之间,平均长度约为1 400 bp,保证了高比例全长cDNA的获得。综合两方面判断,文库质量较高,可用于基因的克隆和筛选,从而为进一步揭示烟草花发育分子机制和烟草遗传改良提供有效途径。
3材料与方法
3.1实验材料
3.1.1烟草材料
以福建烤烟品种翠碧一号为材料,等到现蕾和开花阶段,取下花蕾和花序迅速置于液氮中冷冻,-80℃冰箱保存备用。
3.1.2实验试剂
采用Biospin胶回收试剂盒和质粒小量提取试剂盒,PrimerScript逆转录酶,RNase Inhibitor,SfiI,Ex-Taq,dNTP (10 mmol/L与2.5 mmol/L)、DL2000和DL15000分子标准等均购自大连宝生物公司(TAKARA),连接酶购于NEB公司,大肠杆菌DH5α菌种由本实验室保存,其它试剂为国药集团化学试剂有限公司生产。
3.1.3引物
见表2。
表2 引物名称及序列 Table 1 Primer name and sequence |
3.2试验方法
3.2.1总RNA提取
采用改良的CTAB法提取总RNA:取烟草花材料,加液氮充分研磨呈粉末状,迅速转移到5 mL离心管中,加入2.4 mL CTAB提取缓冲液和2% β-巯基乙醇,迅速振荡混匀,涡旋3 min,65℃水浴约15~20 min,吸取上清液经过两次等量氯仿/异戊醇抽提,去除蛋白后,加入LiCl溶液沉淀两次,用75%乙醇洗涤2次,干燥后分别用RNase free H2O和去离子甲酰胺溶解。得总RNA后,使用ND-2000C微量紫外分光光度计检测总RNA浓度、纯度,1%琼脂糖凝胶电泳检测其完整性。
3.2.2文库构建的主要步骤
主要参照美国Clontech公司开发的SMART文库构建技术进行,根据实验需要,对部分步骤有所改动。即以3 μg总RNA反转录得第一链cDNA,以抑制PCR方法合成双链cDNA,双链经蛋白酶K消化后进行SfiI酶切,将酶切产物经琼脂糖凝胶电泳后,切割回收750 bp以上的区域,再与pDNR-LIB载体连接,电转化大肠杆菌DH5α,获得原始文库。从中随机挑取部分克隆进行菌落PCR和酶切鉴定以确定文库的重组率及插入片段的大小。将原始文库菌液涂板扩增后,得到的菌液按不同比例稀释涂板检测扩增文库滴度。
3.2.3文库初步测序及分析
随机挑取24个单菌落送上海国家人类基因组南方研究中心进行测序,将测序结果去除载体和引物序列后,利用Blast2GO软件进行分析,按照默认的参数 (Blast program: BlastX, Blast database: nr)先运行BlastX,再进行go-mapping和annotation功能注释,其中功能注释主要从分子功能、细胞组件、生物过程三个不同的水平进行,并导出相应的归并数据。
作者贡献
曾建斌、陈华、陈顺辉是本研究的实验设计和实验研究的执行人;曾建斌完成数据分析,论文初稿的写作;张冲和蔡铁城参与部分实验研究;庄伟建是项目的构思者及负责人,指导实验设计,数据分析,论文写作与修改。全体作者都阅读并同意最终的文本。
致谢
本研究是在国烟科(2006)371、闽烟科(2008)13和福建省作物分子与细胞生物学重点实验室项目(2008J1033)共同资助下完成,在此表示感谢。
参考文献
Cong N., Cheng Z.J., and Wang J.M., 2007, The ABCDE model of floral organ development, Zhongguo Nongxue Tongbao (Chinese Agricultural Science Bulletin), 23(7): 124-128 (丛楠, 程治军, 万建民, 2007, 控制花器官发育的ABCDE模型, 中国农学通报, 23(7): 124-128)
Cui R.F., and Meng Z., 2007, Functional conservation and diversity of floral homeotic MADS-box genes in Angiosperms, Zhiwuxue Tongbao (Chinese Bulletin of Botany), 24(1): 31-41 (崔荣峰, 孟征, 2007, 花同源异型MADS-box基因在被子植物中的功能保守性和多样性, 植物学通报, 24(1): 31-41)
Guo Y.L., Zhu Q.L, Zheng S.Y., and Li M.Y., 2007, Cloning of a MADS-box gene (GhMADS3) from cotton and analysis of its homeotic role in transgenic tobacco, Journal of Genetics and Genomics, 34(6): 527-535 doi:10.1016/S1673-8527(07)60058-7
Han L.T., Jiang W., Yang S.P., YU D.Y., and Gai J.Y., 2010, Isolation and analysis of MADS-box gene from Soybean (Glycine max L. Merr.) cytoplasmic male sterile line, Zuowu Xuebao (Acta Agronomica Sinica), 36(6): 905-910 (韩利涛, 姜伟, 杨守萍, 喻德跃, 盖钧镒, 2010, 大豆细胞质雄性不育系MADS-box基因的分离分析, 作物学报, 36(6): 905-910)
Hua Z.M., 1998, The molecular mechanism of floral formation of plants, Zhiwuxue Tongxun (Plant Physiology Communications), 34(1): 74-79 (华志明, 1998, 植物成花分子机理研究的进展, 植物生理学通讯, 34(1): 74-79)
Kramer E.M., Jaramillo M.A., and Stilio V.S.D., 2004, Patterns of gene duplicationand functional evolution during the diversification of the AGAMOUS subfamily of MADS-box genes in angiosperms, Genetics, 166(2): 1011-1023 doi:10.1534/genetics.166.2.1011
Li J.T., Chen P., Li J., Liu P., He Y.Y., and Wang Q.Y., 2010, Construction of a muscle cDNA library of Chinese shrimp Fenneropenaeus chinensis and sequence analysis of the troponin I gene, Journal of Ocean University of China, 9: 81-86 doi:10.1007/s11802-010-0081-1
Lin J.T., Wang C.R., Pramanik J., Zhang H.Y., Feng H.G., Yang B.S., Li Y.C., and Xu C.S., 2004, Study on construction of cDNA library from rat nomal liver and regeneration liver with SMART technique, Indian Journal of Clinical Biochemistry, 19(2): 177-180
Shi T., Gao Z.H., Zhang Z., Zhuang W.B., 2010, Advance of research on microRNA in flower development regulation, Zhongguo Nongxue Tongbao (Chinese Agricultural Science Bulletin), 26(13): 267-271 (侍婷, 高志红, 章镇, 庄维兵, 2010, MicroRNA参与植物花发育调控的研究进展, 中国农学通报, 26(13): 267-271)
Wang J.B., Xu B.Y., Jin Z.Q., and Feng C., 2009, Construction cDNAlibrary of using SMART technology, Redai Zuowu Xuebao (Chinese Journal of Tropical Crops), 30(8): 1109-1112 (王家保, 徐碧玉, 金志强, 冯超, 2009, 利用SMART技术构建荔枝果皮cDNA文库, 热带作物学报, 2009, 30(8): 1109-1112)
Wang T., Ma Y.P., and Dai S.L., 2010, The molecular mechanism in regulation of flowering in ornamental plants,Zhiwu Xuebao (Chinese Bulletin of Botany), 45(6): 641-653 (王翊, 马月萍, 戴思兰, 2010, 观赏植物花期调控途径及其分子机制, 植物学报, 45(6): 641-653)
Zhu B.Y., Gao L., Huang X., and Li H.M., 2009, Construction of Monascus full-length cDNA library with SMART approach, Guangdong Yaoxueyuan Xuebao (Journal of Guangdong Pharmaceutical College), 25(3): 310-313 (朱碧云, 高蓝, 黄欣, 李浩明, 2009, SMART技术构建红曲霉全长cDNA文库, 广东药学院学报, 25(3): 310-313)